GP Nord Ltd
Reg. Nr: 40203177406
Address: Kurzemes prospekts 23, Rīga, LV-1067, Latvija
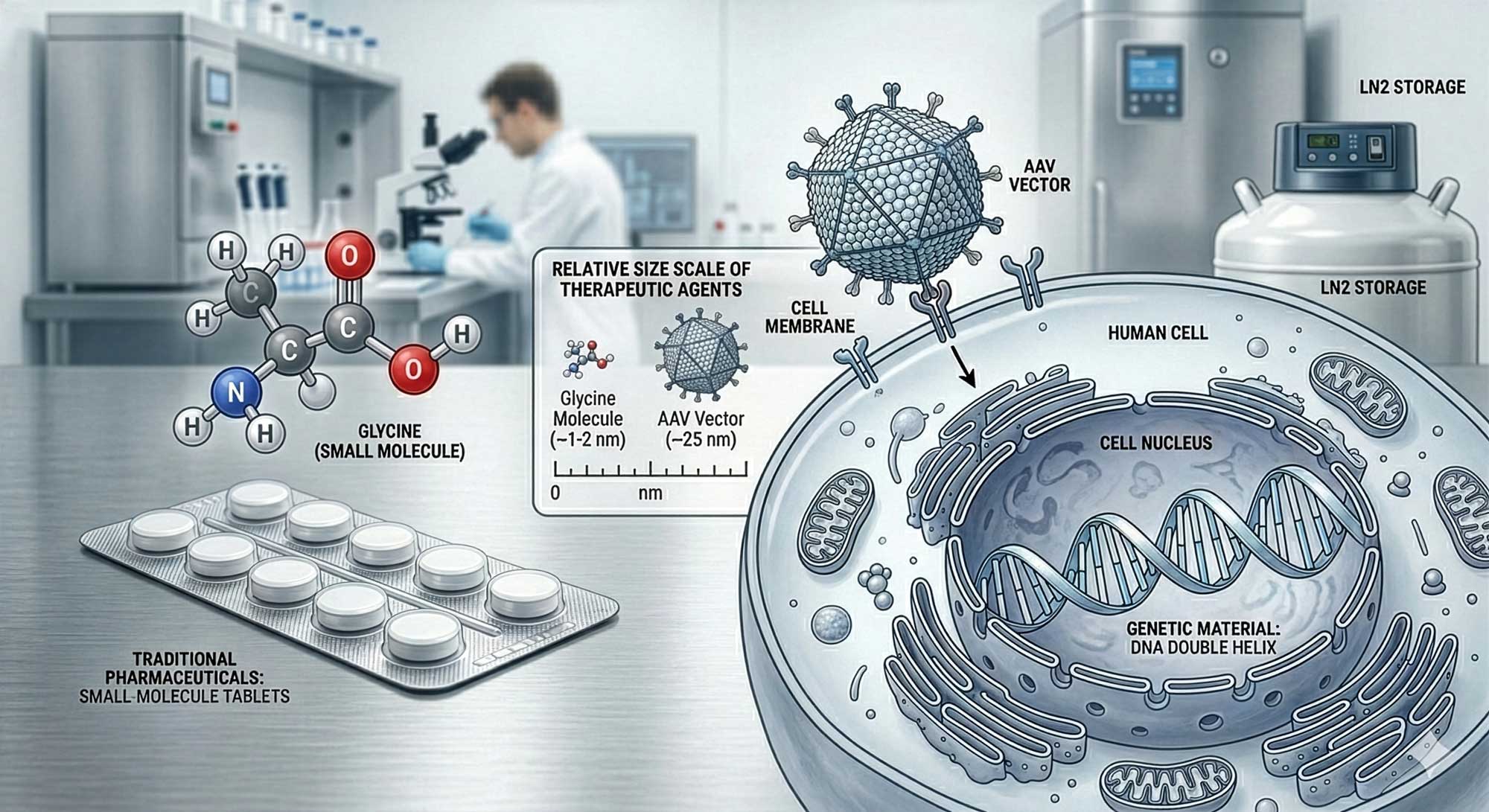
Table of Contents
1. Essay 1: Introduction & Fundamentals of Gene and Cell Therapy
2. Essay 2 — Scientific & Technical State of the Art
3. Essay 3 — Regulatory & Market Context
4. Essay 4 — Manufacturing & Supply Constraints
5. Essay 5 — Outlook: 5–10 Year Evidence-Based Forecast
6. References
Essay 1 – Introduction & Fundamentals of Gene and Cell Therapy
Gene and cell therapies represent a structural shift in how medicine conceptualizes treatment. Instead of modulating disease pathways with small molecules or recombinant proteins, these approaches attempt to modify or replace dysfunctional biological components directly at the genetic or cellular level. For pharmaceutical wholesalers operating in Europe, understanding this field is not optional. It is becoming a defined segment of advanced therapeutics with specific regulatory, logistical, and economic implications.
1. What Is Gene Therapy?
Gene therapy involves the delivery, removal, or editing of genetic material within a patient’s cells to treat disease. The concept dates back to the 1970s, but the first approved gene therapy in Europe was Glybera in 2012, authorized by the European Medicines Agency (EMA). It targeted lipoprotein lipase deficiency, although it was later withdrawn for commercial reasons.
Today, gene therapies most commonly use viral vectors such as adeno-associated viruses (AAV) or lentiviruses to deliver therapeutic DNA. According to the U.S. Food and Drug Administration (FDA), more than 30 gene therapy products have been approved globally as of 2025, including treatments for spinal muscular atrophy and inherited retinal dystrophy. The European Medicines Agency (EMA) classifies gene therapies under the broader category of Advanced Therapy Medicinal Products (ATMPs).
Hard fact 1: Over 2,000 gene therapy clinical trials are registered globally (WHO ICTRP database, 2024).
Hard fact 2: The FDA projected in 2019 that it would be approving 10–20 cell and gene therapies per year by the mid-2020s.
2. What Is Cell Therapy?
Cell therapy uses living cells as therapeutic agents. These may be autologous (derived from the patient) or allogeneic (from donors). The most established example is CAR-T therapy (Chimeric Antigen Receptor T cells), first approved in the EU in 2018.
CAR-T therapies genetically modify a patient’s T cells to recognize and destroy cancer cells. According to IQVIA, CAR-T revenues exceeded USD 4 billion globally in 2023, driven primarily by hematological oncology indications.
Hard fact 3: More than 40 ATMPs have received marketing authorization in the EU since the ATMP regulation entered into force in 2008 (EMA data, 2024).
Hard fact 4: Median list prices for one-time gene therapies in Europe typically range from EUR 250,000 to over EUR 2 million per patient (OECD pharmaceutical pricing reports).
3. Conceptual Differences from Conventional Pharmaceuticals
Gene and cell therapies differ from traditional drugs in at least three fundamental dimensions:
- 1.Mechanism of action — They are often curative or long-acting after a single administration.
- 2.Manufacturing complexity — Production involves biological systems rather than chemical synthesis.
- 3.Distribution model — Some therapies require vein-to-vein logistics within 2–4 weeks.
This has consequences for wholesalers. The model shifts from high-volume SKU management to controlled distribution of ultra-high-value, low-volume products.
4. Clinical Scope
Initially concentrated in rare monogenic disorders, gene therapies are expanding into oncology, hemophilia, sickle cell disease, and potentially cardiovascular and neurodegenerative conditions. The New England Journal of Medicine (NEJM) has published multiple pivotal trials demonstrating sustained remission in hemophilia B following AAV-mediated gene transfer, with factor IX activity maintained over several years.
Hard fact 5: Sickle cell gene therapy demonstrated >90% reduction in vaso-occlusive crises in phase 3 trials (NEJM, 2023).
5. Strategic Relevance for European Distributors
For European pharmaceutical wholesalers, gene and cell therapy is not yet a volume driver but a structural indicator. It introduces:
- Higher regulatory scrutiny (traceability, pharmacovigilance)
- Cold chain and cryogenic transport requirements
- Direct hospital coordination models
- Outcome-based reimbursement contracts in several EU markets
The market remains relatively small compared to traditional biologics. However, it is disproportionately complex relative to its size.
This series will explore scientific maturity, regulatory frameworks, manufacturing bottlenecks, and forward-looking market projections relevant to the European pharmaceutical ecosystem.
Essay 2 — Scientific & Technical State of the Art
If Essay 1 outlined definitions and scope, this section addresses where the science actually stands today. Gene and cell therapy is no longer experimental in principle — but it remains technically constrained in execution. The distinction matters.
1. Vector Technologies: Delivery Is the Core Bottleneck
In gene therapy, delivery systems define feasibility. Most approved in vivo gene therapies use adeno-associated virus (AAV) vectors due to their relatively low immunogenicity and stable episomal persistence. However, AAV has a packaging capacity of approximately 4.7 kilobases — limiting the size of genes that can be delivered.
Hard fact 1: More than 60% of in vivo gene therapy clinical trials use AAV vectors (FDA and WHO trial databases, 2024).
Lentiviral vectors dominate ex vivo approaches (e.g., CAR-T, hematopoietic stem cell therapies) because they integrate into the host genome, enabling durable expression. However, integration carries insertional mutagenesis risk, although safety profiles have improved significantly compared to early 2000s trials (NEJM long-term follow-up studies).
Non-viral systems (lipid nanoparticles, electroporation, CRISPR-based ribonucleoprotein delivery) are advancing, particularly following the validation of lipid nanoparticle technology in mRNA vaccines. Yet their clinical penetration in gene therapy remains limited.
2. Genome Editing: From Replacement to Precision Repair
First-generation gene therapies focused on gene addition. Current research is shifting toward precise genome editing, primarily using CRISPR-Cas systems.
Hard fact 2: In 2023, the first CRISPR-based therapy for sickle cell disease received regulatory approval in the UK and US, marking the first clinical validation of CRISPR editing (FDA, UK MHRA).
CRISPR editing can occur ex vivo (cells edited outside the body and reinfused) or in vivo. Ex vivo editing is currently safer and more controlled, but operationally complex. In vivo editing remains technically challenging due to delivery precision and off-target risks.
Emerging editing platforms include:
- Base editing (single nucleotide changes without double-strand breaks)
- Prime editing (more flexible sequence replacement)
- Epigenetic modulation without DNA cutting
These platforms aim to reduce genotoxicity risks and improve therapeutic durability.
3. Cell Therapy Engineering: CAR-T and Beyond
CAR-T therapy is now established in hematologic malignancies. As of 2024, six CAR-T products are approved in the EU and US (EMA, FDA).
Hard fact 3: Complete response rates in relapsed/refractory B-cell acute lymphoblastic leukemia exceed 80% in pivotal CAR-T trials (NEJM data).
However, scientific limitations persist:
- Cytokine release syndrome (CRS) remains a common toxicity.
- Manufacturing failure rates range between 5–10% depending on indication (IQVIA manufacturing analyses).
- Solid tumor efficacy remains limited due to tumor microenvironment barriers.
Next-generation approaches include:
- Allogeneic “off-the-shelf” CAR-T cells
- CAR-NK (natural killer) cell platforms
- Tumor-infiltrating lymphocyte (TIL) therapies
- Gene-edited universal donor cells
The shift toward allogeneic platforms is scientifically and economically important because it could decouple manufacturing from individual patients.
4. Durability and Long-Term Monitoring
A defining feature of gene therapy is its claim of durability. However, long-term data remain limited for many products.
Hard fact 4: Regulatory authorities typically require 15 years of follow-up for integrating gene therapies due to potential delayed adverse events (FDA guidance on long-term follow-up, EMA ATMP guidance).
Durability varies:
- Hemophilia gene therapies show multi-year sustained factor expression.
- Some CAR-T therapies demonstrate relapse rates within 2–3 years.
- Vector immunity can prevent redosing in AAV-based therapies.
This raises a scientific tension: therapies marketed as “one-time” may require re-intervention if expression declines.
5. Scientific Maturity Level
The field can be described as:
- Technically validated in rare monogenic disorders
- Operationally viable in hematologic oncology
- Experimental in complex polygenic diseases
Gene therapy for neurodegeneration, cardiovascular disease, and autoimmune conditions remains largely investigational.
Hard fact 5: According to ClinicalTrials.gov, over 70% of gene therapy trials are still in Phase I or II (2024 data).
In other words, the pipeline is broad, but late-stage validation remains concentrated in oncology and rare diseases.
6. Implications for Pharmaceutical Wholesalers
From a distribution perspective, the scientific state translates into:
- Small but high-value product portfolios
- Indication concentration in tertiary hospital centers
- Significant pharmacovigilance and traceability requirements
- Uncertainty in long-term demand predictability
The science is credible. The scale is not yet industrial.
In the next essay, we examine how European regulatory systems and market dynamics are adapting to these realities.
Essay 3 — Regulatory & Market Context
Scientific feasibility does not automatically translate into market sustainability. Gene and cell therapies operate within one of the most complex regulatory and reimbursement environments in modern pharmaceuticals. For European wholesalers, the regulatory architecture is not abstract — it determines access pathways, distribution models, and financial exposure.
1. European Regulatory Framework
In the European Union, gene and cell therapies are regulated as Advanced Therapy Medicinal Products (ATMPs) under Regulation (EC) No 1394/2007. The European Medicines Agency (EMA) centrally authorizes all ATMPs via the Committee for Advanced Therapies (CAT) and the Committee for Medicinal Products for Human Use (CHMP).
Hard fact 1: Since 2008, over 40 ATMPs have received EU marketing authorization, though several were later withdrawn for commercial reasons (EMA ATMP database, 2024).
Authorization requires:
- Centralized EMA approval
- Risk Management Plan (RMP)
- Long-term safety follow-up (often 10–15 years)
- Post-authorization studies
Compared to small molecules, regulatory scrutiny is significantly higher, especially regarding traceability and pharmacovigilance.
2. FDA vs EMA: Convergence and Differences
The U.S. Food and Drug Administration (FDA) regulates these products through the Center for Biologics Evaluation and Research (CBER). Both agencies require long-term monitoring and post-marketing commitments, but accelerated pathways (e.g., FDA Breakthrough Therapy Designation, EMA PRIME scheme) are commonly used.
Hard fact 2: Over 70% of approved gene and cell therapies received accelerated or conditional approval pathways (FDA, EMA public records).
The practical implication is earlier market entry but continued evidence generation obligations.
For wholesalers operating across EU markets, understanding conditional approvals is critical — reimbursement negotiations often lag behind marketing authorization.
3. Pricing and Reimbursement Models
Gene therapies are frequently one-time treatments with very high upfront costs. OECD and IQVIA analyses indicate list prices in Europe typically range between EUR 250,000 and EUR 2 million per patient.
Hard fact 3: Zolgensma (gene therapy for spinal muscular atrophy) entered European markets at a list price exceeding EUR 1.9 million per treatment (OECD pharmaceutical pricing review).
Traditional cost-per-unit models are strained by such pricing structures. As a result, European payers increasingly use:
- Outcome-based reimbursement agreements
- Staggered or annuity payments
- Risk-sharing contracts
Italy and Germany have piloted outcome-linked payment schemes for certain gene therapies (OECD, 2023).
However, real-world implementation remains administratively complex. Evidence collection infrastructure varies across Member States.
4. Market Size and Growth
While strategically important, gene and cell therapy remains a small percentage of total pharmaceutical spending.
Hard fact 4: According to IQVIA (2024), advanced therapies represent less than 2% of global pharmaceutical market volume but account for a disproportionately high share of expenditure growth.
Oncology dominates the commercial segment, particularly CAR-T therapies in hematologic malignancies. Rare genetic diseases follow.
Geographically:
- The United States leads in approvals and revenues
- Germany and France are the largest EU markets
- Central and Eastern Europe show slower adoption due to budget constraints
For European wholesalers, this creates uneven demand patterns across regions.
5. Access Inequality
WHO and OECD reports highlight disparities in access to advanced therapies between high-income and middle-income countries.
Hard fact 5: Fewer than 20 countries globally account for the vast majority of gene therapy administrations (WHO policy briefs, 2024).
Within the EU, reimbursement timelines differ significantly:
- Germany often enables earlier access through its AMNOG framework
- Some EU markets experience delays exceeding 12–24 months post-EMA approval
This heterogeneity complicates inventory planning and forecasting.
6. Hospital-Centric Distribution
Unlike conventional biologics, most gene and cell therapies are administered in specialized tertiary centers. Distribution often occurs through controlled direct-to-hospital models rather than broad wholesaler stock circulation.
Key characteristics:
- Low-volume, high-value shipments
- Strict temperature control (cryogenic for CAR-T)
- Patient-specific tracking requirements
- Manufacturing-to-infusion chain coordination
This shifts the role of wholesalers from inventory managers to controlled logistics partners.
7. Structural Observations
The regulatory environment is supportive but cautious. The market environment is innovative but financially constrained.
The core tension:
- Therapies promise long-term value
- Payment systems operate on annual budgets
For wholesalers, gene and cell therapy is less about margin expansion and more about operational capability and regulatory alignment.
In Essay 4, we examine the manufacturing and supply constraints that shape availability and scalability.
Essay 4 — Manufacturing & Supply Constraints
If regulation defines access, manufacturing defines reality. Gene and cell therapies are not constrained primarily by scientific theory or even regulatory approval. They are constrained by production capacity, process variability, and logistics. For European pharmaceutical wholesalers, this is where strategic relevance becomes operational.
1. Manufacturing Is Not Standard Biologics Production
Conventional biologics are produced in large bioreactors with relatively standardized upstream and downstream processes. Gene and cell therapies are fundamentally different.
Autologous CAR-T therapies, for example, require:
- 1.Leukapheresis (cell collection from the patient)
- 2.Transport to a specialized manufacturing site
- 3.Genetic modification (often via lentiviral transduction)
- 4.Expansion and quality control
- 5.Cryogenic shipment back to the hospital
- 6.Patient infusion
This “vein-to-vein” cycle typically takes 2–4 weeks under optimal conditions.
Hard fact 1: Manufacturing turnaround times for autologous CAR-T products range from 16 to 28 days (EMA product dossiers; NEJM clinical reports).
Each batch corresponds to one patient. This eliminates economies of scale typical for small molecules or monoclonal antibodies.
2. Capacity Limitations
Manufacturing capacity remains limited relative to clinical demand growth.
Hard fact 2: IQVIA (2024) estimates that global viral vector manufacturing capacity remains insufficient to meet projected demand if late-stage pipeline products are approved at current rates.
AAV vector production is particularly constrained due to:
- Low yields in HEK293 or insect cell systems
- Complex purification processes
- High rejection rates during quality testing
Viral vector production failure rates in early-stage facilities have been reported in the 10–20% range (industry analyses cited by FDA and EMA advisory discussions).
The capital cost of establishing GMP-compliant viral vector facilities often exceeds EUR 100–300 million depending on scale.
3. Quality Control and Batch Failure
Unlike conventional pharmaceuticals, batch release in gene and cell therapy involves:
- Sterility testing
- Replication-competent virus testing
- Potency assays
- Genetic integrity confirmation
Hard fact 3: CAR-T manufacturing failure rates due to insufficient cell expansion or contamination range between 5–10% depending on indication and manufacturer (IQVIA manufacturing insights, 2023).
In autologous therapies, a failed batch directly affects a specific patient — not anonymous market inventory.
4. Cold Chain and Cryogenic Logistics
Distribution requirements are equally specialized.
CAR-T products are typically transported in vapor-phase liquid nitrogen at temperatures below –150°C. Gene therapies using AAV vectors often require storage at –60°C to –80°C.
Hard fact 4: Deviations in ultra-cold chain conditions can render cell therapy batches unusable, with per-shipment values exceeding EUR 300,000–500,000 (OECD advanced therapy cost analyses).
This necessitates:
- GPS-enabled temperature monitoring
- Validated cryoshippers
- Redundant transport planning
- Real-time coordination between hospital and manufacturer
For wholesalers, this represents a shift from pallet-based cold chain management (2–8°C) to individual shipment orchestration.
5. Allogeneic Platforms: Potential Structural Change
Allogeneic (“off-the-shelf”) cell therapies aim to industrialize production by using donor-derived cells that can be manufactured in larger batches.
Scientifically, this requires:
- Gene editing to reduce graft-versus-host disease risk
- Immune evasion engineering
- Scalable cell banking systems
Hard fact 5: More than 40 allogeneic cell therapy candidates are currently in clinical development globally (WHO ICTRP and ClinicalTrials.gov aggregate data, 2024).
If successful, allogeneic platforms could reduce per-unit costs and simplify logistics — but long-term efficacy data remain limited.
6. European Manufacturing Landscape
Europe hosts several advanced therapy manufacturing hubs, particularly in Germany, the Netherlands, France, and the UK. However, dependence on US-based manufacturing remains significant for certain products.
Supply chain vulnerabilities include:
- Viral vector shortages
- Skilled workforce constraints
- Cross-border regulatory inspections
- Dependence on single-site production models
The COVID-19 pandemic exposed fragility in cross-border pharmaceutical logistics. Advanced therapies amplify that fragility due to low redundancy.
7. Operational Implications for Wholesalers
From a distribution standpoint, gene and cell therapy requires:
- Specialized ultra-cold logistics partnerships
- Integration with hospital scheduling systems
- High-level pharmacovigilance documentation
- Insurance and risk mitigation frameworks
The market is not constrained by demand. It is constrained by production reliability and distribution precision.
In the final essay, we assess the 5–10 year outlook: whether manufacturing scale, regulatory adaptation, and economic sustainability will converge into a mature therapeutic segment.
Essay 5 — Outlook: 5–10 Year Evidence-Based Forecast
Forecasting gene and cell therapy requires separating scientific optimism from structural constraints. The next decade will not determine whether the technology works — that question has largely been answered in specific indications. The real question is scale, sustainability, and integration into national health systems.
1. Pipeline Maturation: From Rare to Broader Indications
The current approved portfolio is concentrated in rare genetic diseases and hematologic oncology. However, the late-stage pipeline is expanding into:
- Hemophilia A and B
- Sickle cell disease
- Beta-thalassemia
- Multiple myeloma
- Selected solid tumors
Hard fact 1: As of 2024, more than 1,800 gene and cell therapy trials are ongoing globally, with over 250 in Phase III (WHO ICTRP, ClinicalTrials.gov aggregated data).
Over the next 5–10 years, approvals are likely to remain concentrated in oncology and hematology. Expansion into complex polygenic diseases (e.g., Alzheimer’s, cardiovascular disease) is unlikely to reach widespread commercialization within this timeframe due to biological complexity and delivery challenges.
2. Regulatory Evolution: Adaptive but Cautious
Regulators are not slowing development but are refining oversight.
The European Medicines Agency (EMA) and FDA increasingly require:
- Long-term follow-up registries
- Real-world evidence collection
- Risk mitigation strategies
Hard fact 2: Both FDA and EMA recommend up to 15 years of post-treatment monitoring for integrating gene therapies (FDA guidance; EMA ATMP framework).
Over the next decade, regulatory harmonization between the EU, US, and UK is likely to improve data-sharing mechanisms, particularly for pharmacovigilance.
Conditional approvals will continue, but confirmatory evidence will determine product longevity. Withdrawal risk remains real if durability claims are not substantiated.
3. Pricing Pressure and Reimbursement Reform
Current price structures are politically sensitive. OECD reports emphasize that one-time therapies exceeding EUR 1 million challenge annualized health budgets.
Hard fact 3: OECD analyses show that high-cost advanced therapies account for a disproportionate share of pharmaceutical expenditure growth despite representing a small patient population (OECD Pharmaceutical Outlook, 2023).
Over the next 5–10 years:
- Outcome-based reimbursement contracts will become more standardized.
- Installment-based payment models will likely expand in Germany, Italy, and France.
- Health Technology Assessment (HTA) frameworks under the new EU HTA Regulation (effective 2025) will centralize clinical evaluation but leave pricing decisions national.
This may improve predictability but not necessarily speed of access.
4. Manufacturing Scale and Technological Shift
Manufacturing is the most significant determinant of market expansion.
Three developments are likely:
- 1.Increased investment in European viral vector capacity
- 2.Gradual shift toward allogeneic cell platforms
- 3.Automation of cell processing to reduce variability
Hard fact 4: Industry investment in advanced therapy manufacturing facilities in Europe exceeded several billion EUR cumulatively between 2020–2024 (EMA and industry reports).
If allogeneic platforms achieve durable efficacy comparable to autologous CAR-T, cost per treatment could decline meaningfully. However, clinical validation remains ongoing.
AAV vector redosing limitations and pre-existing immunity remain scientific constraints that may limit broad population use.
5. Market Size Projection
IQVIA projects advanced therapies to represent a mid-single-digit percentage of global pharmaceutical spending by the early 2030s.
Hard fact 5: Global advanced therapy revenues are projected to exceed USD 60–80 billion annually by 2030 under moderate growth scenarios (IQVIA Institute, 2024 outlook).
Even under optimistic scenarios, this remains smaller than traditional oncology biologics. Gene and cell therapy will likely remain high-value but low-volume relative to overall pharmaceutical trade.
6. Structural Implications for European Wholesalers
The next decade will not transform wholesalers into gene therapy manufacturers. However, it will require:
- Specialized ultra-cold logistics infrastructure
- Data integration with hospital networks
- Strong pharmacovigilance and traceability systems
- Financial risk management for high-value shipments
Distribution models will remain hospital-centered, with limited retail exposure.
The likely equilibrium by 2035:
- Stable but niche therapeutic segment
- Expanded oncology and hematology coverage
- Increased regulatory data transparency
- Moderately improved manufacturing efficiency
- Persistent budget scrutiny
Gene and cell therapy will not replace conventional pharmaceuticals. It will coexist as a technically sophisticated, tightly regulated segment addressing specific high-burden diseases.
For European pharmaceutical wholesalers, strategic preparation is prudent. Overinvestment based on speculative growth is not.
References
- European Commission. Regulation (EC) No 1394/2007 on Advanced Therapy Medicinal Products.
- European Medicines Agency (EMA). Advanced Therapy Medicinal Products (ATMP) overview.
- European Medicines Agency (EMA). ATMP regulatory framework and authorization data.
- European Medicines Agency (EMA). ATMP regulatory framework and post-authorization monitoring guidelines.
- European Medicines Agency (EMA). CAR-T product assessment reports.
- European Union. Regulation (EU) 2021/2282 on Health Technology Assessment.
- IQVIA Institute for Human Data Science. Global Oncology Trends Report. 2023.
- IQVIA Institute for Human Data Science. Advanced Therapy Manufacturing Insights. 2023–2024.
- IQVIA Institute for Human Data Science. Global Trends in Advanced Therapies. 2024.
- New England Journal of Medicine (NEJM). Gene therapy trials in hemophilia and sickle cell disease. 2023.
- New England Journal of Medicine (NEJM). CAR-T manufacturing and clinical outcome publications.
- New England Journal of Medicine (NEJM). CAR-T and gene editing clinical trial publications. 2022–2024.
- OECD. Pharmaceutical Pricing Policies in a Global Market.
- OECD. Pharmaceutical Pricing and Reimbursement Policies. 2023.
- OECD. High-Cost Medicines and Advanced Therapy Pricing Analysis.
- OECD. Pharmaceutical Outlook 2023: High-Cost Medicines.
- U.S. Food and Drug Administration (FDA). Statement on regenerative medicine framework. 2019.
- U.S. Food and Drug Administration (FDA). Long-Term Follow-Up After Administration of Human Gene Therapy Products.
- U.S. Food and Drug Administration (FDA). CBER Annual Reports.
- U.S. Food and Drug Administration (FDA). CMC guidance for human gene therapy products.
- UK Medicines and Healthcare products Regulatory Agency (MHRA). CRISPR therapy approval announcement. 2023.
- World Health Organization (WHO). International Clinical Trials Registry Platform (ICTRP). 2024.
- World Health Organization (WHO). Access to Advanced Therapies Policy Brief. 2024.